Recently, a research team led by Prof. CHEN Ping, Prof. GUO Jianping and Associate Prof. WANG Qianru from Dalian Institute of Chemical Physics (DICP) has applied hydride materials to catalytic selective hydrogenation reactions, achieving chemoselective hydrogenation of quinolines and stereoselective hydrogenation of internal alkynes, respectively.
Selective hydrogenation reactions have long been regarded as an exclusive research domain of d-block metal catalysts. The research team has verified that hydrides can serve as high-efficiency catalysts for selective hydrogenation, exhibiting a working principle distinct from that of conventional transition metal catalysis. These findings provide a different strategy for developing catalytic materials and reaction mode beyond transition metal systems.
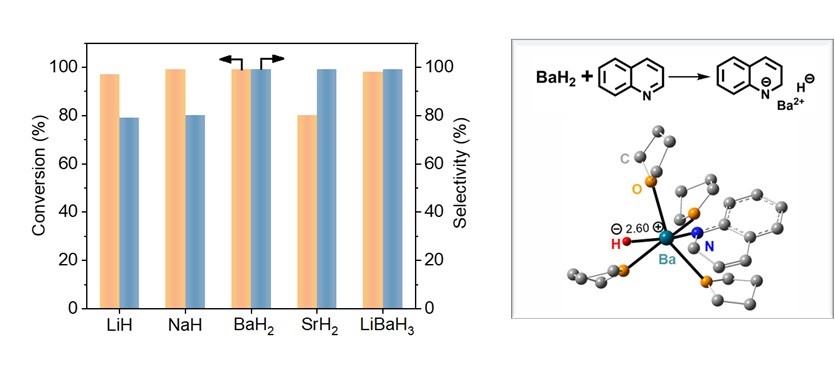
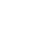
In the hydrogenation of quinoline, it has been found that alkaline and alkaline-earth metal hydrides, such as lithium hydride (LiH), sodium hydride (NaH), barium hydride (BaH2), strontium hydride (SrH2) and lithium barium hydride (LiBaH3), can efficiently catalyze the selective hydrogenation of quinoline to 1,2,3,4-tetrahydroquinoline under relatively mild conditions. Among them, BaH2 delivers the most prominent catalytic performance, comparable to state-of-the-art non-noble metal catalytic systems based on iron, cobalt and nickel reported in recent years. Compared with transition metal catalysis, this strategy features simple operation without the requirement of sophisticated ligands or co-catalysts. Mechanistic investigations reveal that the in-situ generated soluble barium hydride active species are the key to efficient chemoselective hydrogenation, which fundamentally differs from transition metal catalytic systems in substrate coordination and the activation of C=C and C=N bonds.
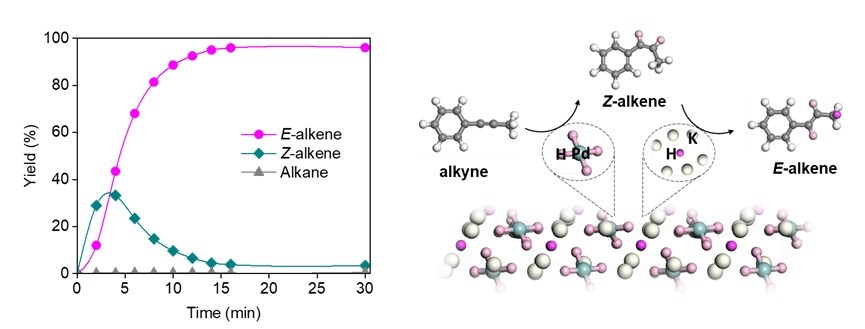
For the hydrogenation of internal alkynes, the ternary hydride K3PdH5 has been demonstrated to efficiently catalyze the tandem semi-hydrogenation–isomerization reaction of internal alkynes, affording trans-alkene products with high stereoselectivity. The reaction rate is two orders of magnitude higher than that of existing catalytic systems. Mechanistic studies indicate that K3PdH5 realizes atomic-level coupling of semi-hydrogenation active sites [Pd–H] and isomerization active sites [K–H] within a single lattice, optimizing mass and energy transfer between the two types of active sites. This work offers a new route for the rational design of high-performance heterogeneous tandem catalysts.
Article link: https://doi.org/10.1039/D5SC07195J; https://doi.org/10.1021/jacs.6c00931